In-Cell Western Assay Development Project
Assay Development Workflow Overview
The assay development steps shown below are intended to give you a general idea for how an In‑Cell Western Assay can be developed.
Generally, the steps of assay development can follow the steps in the protocol (see Example Protocol Overview). In practice, the development process is iterative, and steps may need to be repeated. It is up to each researcher to develop an assay that is fit for their purposes.
At any point during the assay development process, you can use a Z'-Factor assay to evaluate the robustness of the assay under development.
Preset Plate Templates (see the [Preset] Z-Factor Determination Plate Templates) are provided in Empiria Studio® Software to help you get started.

Step 1. Start by Defining Your Experimental System
Although your project will have specific questions, here are some common and important questions to consider.
What target do you want to detect?
What antibodies are available to detect your target?
Is the target on the cell surface or intracellular?
What cell line(s) should you use?
What controls should you use?
Which imager will you be using for detection?
Step 2. Determine the Best Normalization Method for Your Assay
Normalization is the process of minimizing how much affect cell number variation between wells will have on your results. Because some variation is unavoidable, normalization is essential for an In‑Cell Western Assay.
Read Normalization or see the other steps in this section for more information about how normalization affects other parts of assay development.
Step 3. Determine the Cell Number to Seed
Determine the linear range of detection for your normalization method and target. The appropriate cell number should be within the linear range of detection and result in cells that are between 70 to 80 percent confluent at the time of detection.
The Plate Analysis Template [Preset] Cell Stain Linearity is provided in Empiria Studio® Software to help you analyze or design your experiment for determining the cell number to seed in your assay.
Step 4. Validate Antibodies
Research which antibodies are available to detect your target. The Antibody Selection chapter (page Antibody Selection) has recommendations for researching antibodies to use in your assay. A Western blot, In‑Cell Western, or immunofluorescence assay could be used to validate antibodies.
In a Western blot validation experiment, ensure there are no unexpected bands. Empiria Studio Software provides workflows to help with antibody validation for Western blots and antibody titration for In‑Cell Western assays.
Step 5. Choose Fixation and Permeabilization Conditions
Choose a variety of fixation and permeabilization conditions to evaluate, and run an In‑Cell Western to determine the best conditions for your assay.
Start with vendor recommendations, if available. If your assay requires multiple antibodies that have differing vendor recommendations, you may need to choose the conditions that work best overall instead of using conditions that are optimal for just one of the antibodies.
The [Preset] Fixation and Permeabilization Evaluation Plate Template can be used to determine the optimal fixation and permeabilization conditions for an experiment.
Step 6. Choose a Blocking Buffer
Choose a variety of blocking buffers to evaluate, and run an In‑Cell Western to determine the best blocking buffer for your assay.
Start with vendor recommendations, if available. If your assay requires multiple antibodies that have differing vendor recommendations, you may need to choose the conditions that work best overall instead of using conditions that are optimal for just one of the antibodies.
You can analyze In‑Cell Western blocking buffer experiment results using these Plate Templates provided in Empiria Studio® Software: [Preset] Blocker Evaluation (Single Antibody) and [Preset] Blocker Evaluation (Two Antibodies).
Step 7. Determine Primary Antibody Concentration
Run an Antibody Titration experiment to determine the antibody concentration that provides the largest fold change between positive and negative controls and minimal background.
Try starting with the dilution given on the pack insert for IF and include a dilution that is two-fold above and one that is two-fold below. Follow dilution recommendations on the pack insert for IRDye® Secondary Antibodies.
Two Plate Templates are provided in Empiria Studio Software to help you design and analyze Antibody Titration experiments. The two Plate Templates are [Preset] Antibody Titration (Single Antibody) and [Preset] Antibody Titration (Two Antibodies).
Define Experimental System
The first step is define your experimental system. The experimental system for an In‑Cell Western Assay is unique to each project, so this section will broadly cover concepts related to choosing elements of your experimental system, such as choosing replicates and choosing experimental controls.
Replicates
The In-Cell Western Assay is a higher throughput assay than a Western blot. Technical replicates A test performed on the same sample multiple times to establish the variability of a protocol or assay and to determine if an experimental effect is large enough to be distinguished from noise. and biological replicates A test performed on biologically distinct samples representing an identical time point or treatment, used to control for biological variation and to determine if the experimental effect is biologically relevant. can be performed in a single plate, which eliminates the inter-membrane variability associated with Western blots.
Replicates improve the reproducibility and accuracy of experimental findings. They are important because they confirm the validity of observed changes in a biological sample, such as protein levels. Without replication, it is impossible to know if an effect is real or simply an artifact of experimental noise or variation, which can directly affect conclusions made about experimental findings.
There are two types of replicates: biological and technical. Each type addresses different questions (1 - 3). Peer-reviewed journals, such as the Journal of Biological Chemistry and Nature, have specific guidelines regarding replicates.
Clearly define replicates. How many technical and biological replicates were performed during how many independent experiments?
- The Journal of Biological Chemistry, Collecting and Presenting Data (6)
The number of sampled units, N, upon which each reported statistic is based must be stated.
- Nature, Instructions for Authors of Research Articles (Initial Submission) (7)
Technical and Biological Replicates: Which Do You Need to Include?
Technical Replicates
Technical replicates are repeated measurements used to establish the variability of a protocol or assay and determine if an experimental effect is large enough to be reliably distinguished from the assay noise (1). Examples may include loading multiple wells with each sample on the same plate, running multiple plates in parallel, or repeating the plate with the same samples on different days.
Technical replicates evaluate the precision and reproducibility of an assay, which determine if the observed effect can be reliably measured using the assay. When technical replicates are highly variable, it is more difficult to separate the observed effect from the assay variation. You may need to identify and reduce sources of error in your protocol to increase the precision of your assay. Even though technical replicates evaluate the precision and reproducibility of an assay, they do not address the biological relevance of the results.
Biological Replicates
Biological replicates are parallel measurements of biologically distinct and independently generated samples, used to control for biological variation and determine if the experimental effect is biologically relevant. The effect should be reproducibly observed in independent biological samples. Demonstration of a similar effect in another biological context or system can provide further confirmation. Examples include analysis of samples from multiple batches of independently cultured and treated cells.
To demonstrate the same effect in a different experimental context, the experiment might be repeated in multiple cell lines, in related cell types or tissues, or with other biological systems.
An appropriate replication strategy should be developed for each experimental context. Several recent papers discuss considerations for choosing technical and biological replicates (1 - 3).
Technical and Biological Replicates: How Many Replicates Do You Need to Include?
Technical Replicates
Technical replicates are the measurement of the same sample within an assay multiple times. Increasing or decreasing the number of technical replicates can affect the experimental outcome of a research study. A general rule of thumb for plate-based assays is ‘the more replicates the better.’ LICORbio recommends 4 - 6 technical replicates for a 96-well plate. The coefficient of variation increases as well size and working volume decrease. Therefore, for a 384-well plate, a minimum of 4-8 replicates is suggested.
There are several advantages to using many technical replicates within an assay:
Variation can be measured between replicates and statistical tests applied to evaluate differences between the samples.
Smaller changes can be discerned between samples when the average across replicates is calculated.
A larger number of replicates makes identification of outlier samples easier to detect.
Standard Deviation versus Standard Error of the Mean
Standard Deviation
When performing three or more technical replicates, the most important statistical calculation is standard deviation. Standard Deviation (SD) is a measure of the variability of data points with respect to the mean. SD indicates the variation of data within individual samples.
Standard Error of the Mean
The Standard Error of the Mean (SEM) is also sometimes used, although caution should be employed when using the SEM because it is easy to misrepresent variability. SEM compares the means of populations and gives information about the variation in two samples. This information can be valuable to researchers in some circumstances, such as when trying to compare two different experimental groups A set of samples in an experiment (e.g., controls, replicates, treatments) that serve the same purpose in the experimental design.. For example, a cell line treated with an inhibitor versus an untreated cell line.
Biological Replicates
Biological replicates are biologically distinct samples measured in parallel that capture random biological variation. For statistically significant measurements, a minimum of three biological replicates is recommend for plate-based assays in either a 96-well or 384-well format.
Replicates References
1. Naegle, K., Gough, N.R., and Yaffe, M.B. (2015). Criteria for biological reproducibility: what does “n” mean? Sci Signal, 8 (371), fs7. https://doi.org/10.1126/scisignal.aab1125
2. Blainey, P., Krzywinski, M., and Altman, N. (2014). Replication: Quality is often more important than quantity. Nat Meth, 11(9), 879-880. https://www.nature.com/articles/nmeth.3091.pdf
3. Vaux, D.L., Fidler, F., and Cumming, G. (2012). Replicates and repeats – what is the difference and is it significant? EMBO reports, 13(4) 291-296. https://dx.doi.org/10.1038%2Fembor.2012.36
4. Nagele, P. (2003). Misuse of standard error of the mean (SEM) when reporting variability of a sample. A critical evaluation of four anaesthesia journals. Br J Anaesth, 90(4), 514–516. https://doi.org/10.1093/bja/aeg087
5. Lee, D.K., In, J., and Lee, S. (2015). Standard deviation and standard error of the mean. Korean J Anesthesiol, 68(3), 220-223. https://doi.org/10.4097/kjae.2015.68.3.220
6. Journal of Biological Chemistry. Collecting and Presenting Data, (Acc. August 27, 2020).
7. Nature. Instructions for Authors of Research Articles (Initial Submission), (Acc. August 27, 2020).
Experimental Controls
The breadth and power of the In-Cell Western Assay is highly dependent on the chosen antibodies, optimization of each step, and careful consideration and selection of controls. Accurate experimental analysis relies on the framework of data established by the controls.
Control Types and Well Types
The following points describe typical controls for an In‑Cell Western Assay.
Blank control wells contain only PBS buffer and are used to mitigate the potential impact edge effects can have on wells located near the outside of plate.
Secondary antibody background control wells contain secondary antibody without primary antibody or normalization reagent. This control accounts for signal caused by binding of a secondary antibody to material other than the target. By eliminating the primary antibody, any binding of secondary antibody caused by cross-reactivity to cellular antigens or to charged groups created during the fixation process is exposed. If you are using more than one primary antibody, this control will also differentiate secondary antibody binding to its appropriate primary versus cross-reactivity between the multiple secondary antibodies. This control should be included on every plate.
Positive control wells contain primary and secondary antibody. Depending on your experiment, positive controls could be an over-expressed cell line, a treatment known to stimulate target expression, etc.
Negative control wells contain primary and secondary antibody. Depending on your experiment, negative controls could be a CRISPR knockdown, a treatment known to inhibit target expression, etc.
Normalization
In the context of an In-Cell Western Assay, normalization refers to a method of correcting for small differences in the number of cells between wells. Because some variation in cell number is unavoidable, normalization is a critical part of attaining reproducible In-Cell Western data.
This section explains why normalization is necessary, which normalization methods are available, and how to use these methods to get accurate data from your In-Cell Western Assay.
Why Normalization is Necessary
Ideal World
Imagine an In-Cell Western Assay where each well in the plate contained exactly the same number of cells. Normalization would not be necessary for this imaginary experiment.
If you were to add a cell stain to the wells that was known to produce a signal proportional to the number of cells in the well, then the cell stain signal measured for each replicate well would be the same.
The signal measured for a target protein might be the same in each well, or it might differ based on your experimental treatments or conditions.
Either way, you would be sure that the variation in target signal was due to changes in target levels and not the number of cells.

Real World
However, in the real world, the total number of cells will vary, and the target protein levels can also vary. Therefore, normalization is necessary to account for the different cell numbers and to obtain accurate In-Cell Western results.

Normalization Basics
Normalization Concepts
Normalization In an In-Cell Western Assay, normalization means the use of an internal control to mathematically correct for small, unavoidable differences in the number of cells between wells. refers to the use of an internal control Endogenous reference protein(s) that are present in all samples at a stable level and are unaffected by experimental conditions. Signal from detection of a reliable IC should be directly proportional to the number of cells in a well. to mathematically correct for small, unavoidable differences in the number of cells between wells.
An internal control (IC) refers to endogenous reference proteins that are present in all samples at a stable level and are unaffected by experimental conditions. Signal from detection of a reliable IC should be directly proportional to the number of cells in a well.
An internal control can mean a couple different things.
If you are normalizing p-ERK1 to pan-ERK1, pan-ERK1 is the internal control.
If you are using a cell stain for normalization, "internal control" refers more abstractly to all the biomolecules that the cell stain binds. See Cell Stain Strategies for more information.
Normalization Cannot Account for Everything
Normalization is necessary to account for unavoidable cell number differences. Excessive cell number differences must be minimized, because normalization cannot correct for all types and degrees of variation.
Sources of Variability That Normalization Can Correct
Pipetting inconsistency and edge effects can introduce variation from well to well. Normalization can account for small differences. For normalization to be effective, it is important to quantify cell number before seeding to seed cells as accurately as possible.
Sources of Variability That Normalization Cannot Correct
Normalization cannot account for the following possible sources of error.
Fixation/Permeabilization: Normalization cannot compensate for variability introduced during the fixation/permeabilization process.
Inconsistent or inaccurate technique: Care must be taken to ensure accuracy and consistency in lab technique.
Normalization Strategies
There are a couple of primary normalization methods that can be used for In-Cell Western Assays. The different methods either rely on the detection of a constitutively expressed protein or a cell stain. The following methods may work better in some In-Cell Western experiments than in others. It is up to the researcher to determine which strategy is appropriate for given experimental conditions.
Cell Stain Strategies
A cell stain A cell stain (or whole cell stain) is a label that binds to components of a cell to provide a signal readout that is proportional to the number of cells in a sample for normalization. is a reagent that binds to cells so extensively that signal detected from the cell stain proportionately represents the number of cells in the well. A cell stain can be used as a tool for normalization. Because cell stains target different classes of biomolecules within a cell (e.g., proteins and/or nucleic acids), error and variability are minimized. In general, LI-COR recommends using cell stain normalization.
CellTag™ Stain
LICORbio offers a solution for In‑Cell Western Assay normalization using the CellTag Stain (i.e., CellTag 520 Stain and CellTag 700 Stain). CellTag stains accumulate in both the nucleus and cytoplasm of permeabilized cells, and provide a linear fluorescent signal across a wide range of cell types and cell numbers.
CellTag 700 Stain is detected in the 700 nm channel of Odyssey Imagers and CellTag 520 Stain is detected in the 520 nm channel (available on the Odyssey M Imager), allowing accurate quantification of target protein levels when combined with an IRDye® 800CW Secondary Antibody. See licor.com/celltag for more product information.
CellTag™ Stain Stain for the In‑Cell Western Assays
CellTag Stain requires permeabilization, so it cannot be used for normalization in live cells. CellTag Stain is applied to the cells during the IRDye Secondary Antibody incubation step, enabling accurate quantification of target protein levels in an assay with much higher throughput than Western blotting.
CellTag Stain for On-Cell Western Assays
On-Cell Western assays are similar to In‑Cell Western Assays, with the notable difference that the target is detected on the surface of the cell. In some cases, researchers have detected a target on the surface of a cell, then permeabilized the cells and used CellTag Stain for normalization.
Example Data

Sapphire700 and DRAQ5
Sapphire700™ Stain and DRAQ5™ Stain are cell staining agents that can be used in combination for accurate normalization across the same range of cell densities as CellTag 700 Stain or CellTag 520 Stain.
Simultaneous staining of cells with Sapphire700 and DRAQ5 expands the linear range, allowing more accurate normalization of cell number across both low-and high-cell densities. These stains are detected in the 700 nm channel of Odyssey Imagers, allowing accurate measurement of target protein levels in the 800 nm channel when combined with an IRDye® 800CW Secondary Antibody.
Sapphire700 Stain is a non-specific cell stain that accumulates in both the nucleus and cytoplasm of fixed or dead cells, but not live cells. When used to stain serial dilutions of A431 cells in 96-well plates, Sapphire700 displays linearity of fluorescent signal for higher cell densities, from ~50,000 to ~250,000 cells/well.
DRAQ5 Stain is a cell-permeable DNA-interactive agent which can be used for stoichiometric staining of DNA in live or fixed cells. For more information about DRAQ5 Stain, please visit the Biostatus Limited web site (biostatus.com). When serial dilutions of A431 human epithelial carcinoma cells are plated in 96-well plates, DRAQ5 Stain alone demonstrates linearity of fluorescent signal for lower cell densities, from ~3,000 to ~50,000 cells/well in the 700 nm channel for normalization.
Cell Stain Selection Guide
There are many things to consider when selecting the right stain for your application. Below is a list of some questions to ask when choosing the correct stain for your application.
What is the excitation and emission spectra for the stain?
Does your stain work for your organism and/or cell type of interest?
What is the cellular target and/or mechanism of action?
Is the stain compatible with your fixation and permeabilization methods?
Is the stain cell permeable?
Does the dye stain live cells? Dead cells? Or both?
Is the detection range compatible with your experimental design?
Selection Guide for LI-COR Supported Dyes
Stain | Ex/Em (nm) | Detection Range (cells/well) * | Cell Permeable | Localization | Comment |
CellTag™ 700 Stain | 675/697 | 200 – 100,000 | No | Nucleus/Cytoplasm | Wide linear range Cost-effective Easy to use |
Sapphire700™ Stain/DRAQ5™ | 675/697 | 3,000 – 250,000 | No | Nucleus/Cytoplasm | Requires two stains Easy to use |
IRDye® NHS Esters | 689/700 773/792 | 200 – 200,000 | No | Nucleus/Cytoplasm If cells are not permeabilized, the cell surface will be stained. If cells are permeabilized, the nucleus and cytoplasm will be stained. | Use for On-Cell Westerns and In-Cell Westerns Provides flexibility for which channel you use for normalization Requires two additional steps Cost-effective Wide linear range High sensitivity at lower cell numbers |
* The number of cells you use in your experiment will depend on your experimental conditions (experimental conditions can change size/morphology of cells) and the cell line that you are using.
Post-Translational Modification Normalization Strategy
The post-translational modification strategy (PTM) is used to monitor changes in post-translational modification of proteins by normalizing a specific modification of a target protein against all target protein, regardless of modification. The target protein is used as its own internal control.
When using the PTM normalization strategy, you are able to answer the question, “How much does a specific protein modification change compared to the total amount of the protein?”
PTM normalization can be used for normalization in the study of post-translational modification, where suitable antibodies are available.
PTM normalization employs two primary antibodies raised in different hosts:
Modification-specific antibody: An antibody against a specific modification of the target.
Pan-specific antibody: An antibody that recognizes all target regardless of modification.
Spectrally distinct fluorescent secondary antibodies are used to detect the modification-specific and pan-specific antibodies at the same time in the same well (multiplex fluorescent detection).
Validating a PTM Normalization Method
Consider If Epitope Interference Could Be a Problem
Epitope interference may occur if antibodies against different epitopes on the same target interfere with each other's ability to bind the target. If the two antibodies interfere with each other's ability to bind, normalization will not be accurate. Check antibody datasheets to ensure that the two primary antibodies recognize different regions of the protein. The Antibody Publication Database can help you find antibody pairs that work for your experiment (licor.com/antibodyrequest).
Polyclonal pan-specific antibodies are less likely to experience epitope interference. Even if one epitope is blocked, the polyclonal antibody may be able to bind other epitopes.
Depending on the antibodies you are using, you may wish to verify empirically that epitope interference is not occurring.
For example, in 2015, Bakkenist et al. (8) demonstrated a Western blot method that can be used to evaluate binding interference between primary antibody pairs. Identical blots were incubated with modification and pan-specific antibodies separately (singleplex), or with both antibodies simultaneously (multiplex). In that experiment, relatively little variation in signal intensity was measured between pan-specific antibody from multiplex and singleplex blots.
The Western blot method for evaluating binding interference between primary antibody pairs can be adapted to an In‑Cell Western Assay format.
Document Validation Procedure and Results
Document your normalization procedure and results to ensure that all experimental details will be available for publication or reproduction of your results. Include lot numbers of all reagents. New lots of the same catalog number should be verified.
In-Cell Western Protocol with Post-Translational Modification Normalization
After ensuring that your normalization procedure will produce accurate results for your particular experiment, follow the standard protocol for an In-Cell Western Assay to detect the modification-specific and pan-specific proteins in the same well.
Normalization References
8. Bakkenist, C.J., Czambel, R.K., Hershberger, P.A., Tawbi, H., Beumer, J.H., and Schmitz J.C. (2015). A quasiquantitative dual multiplexed immunoblot method to simultaneously analyze ATM and H2AX Phosphorylation in human peripheral blood mononuclear cells. Oncoscience, 2(5), 542–554. https://doi.org/10.18632%2Foncoscience.162
9. Hoffman, G., et al. (2008). A functional siRNA screen for novel regulators of mTORC1 signaling. Poster presentation, American Society for Cell Biology Annual Meeting.
Experiment Linearity
During the development of an In‑Cell Western Assay, it is important to ensure that the signal intensity for both the normalization method and target are each detected within their linear range. Finding a linear range for your target will increase confidence in the assay, but finding this linear range requires many assay-specific considerations. For example, cell may express your target at different levels at different cell densities. Ultimately, it is the researcher's decision how to handle the question of linearity for target detection.
Determining Cell Stain Linearity in Empiria Studio® Software
The following section explains the unique functionality in Empiria Studio Software for determining linear range.
Creating a Plate Template
To determine the cell stain linearity for an In‑Cell Western Assay, first design a Cell Stain Linearity experiment, and set up a Plate Template (with the Cell Stain Linearity category) in the Empiria Studio Experiment Designer. The Plate Template serves dual roles: It is a visual way to plan your experiment, and it is also used for analysis.
![Screenshot of the [Preset] Cell Stain Linearity Plate Template provided in Empiria Studio](../../../Resources/Images/software/empiria_studio/preset-plate-template-cell-stain-linearity-layout.png)
Analyzing Cell Stain Linearity Experiment to Determine Linear Range
In a multiwell plate analysis workflow in Empiria Studio, you choose a multiwell plate image and a Plate Template. The wells designated in the Plate Template are quantified and analyzed.
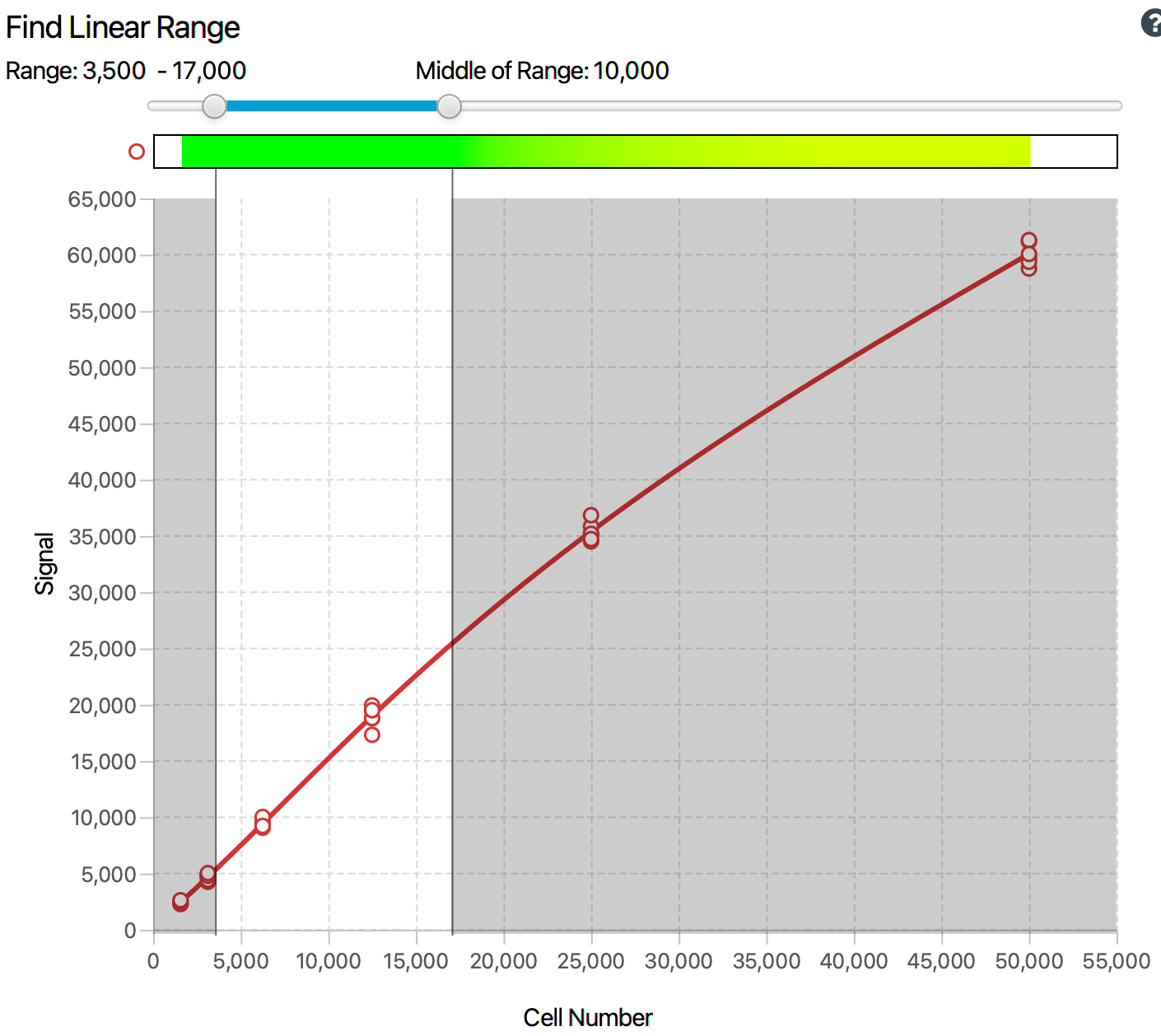
In the Cell Stain Linearity workflow, wells are quantified and the relationship between cell number and signal intensity is graphed on the Find Linear Range page. A color bar above the graph indicates where the relationship between cell number and signal intensity has excellent linearity, good linearity, or no linearity.
Green sections on the bar correspond to regions on the graph where there is excellent linearity between cell number and signal intensity.
Yellow sections on the bar correspond to regions on the graph where there is good linearity.
Red sections on the bar correspond to regions on the graph where the relationship between cell number and signal intensity is not linear.
Drag the slider bars above the graph so that they bracket a region with linearity. The median cell number within this region is indicated above the graph. In general, this is a good cell number to use in your assay to stay within the linear range; however, it is the responsibility of the researcher to choose the appropriate cell number to use.
Antibody Selection
Selection and validation of primary and secondary antibodies are vital to achieving reliable results from an In‑Cell Western Assay.
In this guide, we present antibody validation strategies, examples, and guidelines to ensure accurate and reproducible data. The approach is focused on achieving scientific rigor and lab economics for the In‑Cell Western Assay, but the underlying best practices are applicable to other cell-based assays. Guidelines presented in this document have been written to align with the thorough guidelines provided by the European Antibody Network (euromabnet.com/guidelines).
For a summary of useful resources, see "Additional Resources".
Concept References
To begin, this section explains critical concepts and defines critical terms that will be referenced throughout this section.
Definitions
A detailed glossary of key terms is provided at the end of this document Glossary, but it is worth emphasizing some important definitions that will be used in this section. In particular, it is worth clarifying how "specificity" and "selectivity" will be used.
This section relies on the following definitions from the Workshop Report from the Antibody Validation: Standards, Policies, and Practices Workshop hosted by the Global Biological Sciences Institute (11).
- Affinity: Binding strength between the antibody’s binding site and its epitope.
- Antigen: A substance stimulates the production of and binds to antibodies.
- Epitope: A part of the antigen to which the antibody directly binds.
- Selectivity: Ability to preferably bind the target antigen in the presence of other antigens.
- Specificity: Ability to recognize the target antigen.
Distinguishing Specificity and Selectivity

Identify Assay Requirements
When beginning a new In-Cell Western Assay, identify these basic experimental requirements before acquiring antibodies and completing preliminary method development.
Get to Know Your Target Antigen
To identify an antibody that is both specific and selective for your target The protein you are interested in studying., you will want to acquire a thorough knowledge of your target antigen. In addition to a comprehensive literature review, online databases can be useful resources when searching for information regarding your target antigen. A list of helpful resources is included on page Antibody Resources.
Know your antigen by name(s)
Start your research by identifying the various nomenclature for your antigen to help identify all the relevant information and reagents. The HUGO Gene Nomenclature Committee site (genenames.org) enables you to search human gene nomenclature by symbol, keyword, and ID.
Know your antigen’s sequence(s)
Locate the protein sequence of the antigen, including any known variants (e.g., splicing isoforms, proteolytic products, and post-translational modifications). The UniProt protein database (uniprot.org) is a useful tool for determining your antigen's sequence. Knowing the protein sequence is helpful for making some key decisions.
Is it important to detect all variants of the target antigen or only one in particular?
Is it critical to distinguish domains with different subcellular localizations (e.g., labeling the extracellular versus intracellular portions of membrane protein)?
When variants of a target antigen are present in a sample, you may observe unexpected cellular localization or tissue localization using IF or unexpected bands on a Western blot. Knowing that variants of the target antigen are present may prevent misidentifying non-specificity of the antibody.
Know your antigen’s relatives
It is also valuable to investigate any sequence homology between your target antigen and any related and unrelated proteins. A Basic Local Alignment Search Tool (BLAST) search is useful for identifying these regions of similarity by comparing your target sequence against a large database (blast.ncbi.nlm.nih.gov/Blast.cgi). This search can help you identify proteins that share sequence identity with your target antigen and could potentially bind your antibody. A BLAST search can also identify sequence conservation across species, if you want to utilize a monoclonal antibody against a common epitope found in multiple species.
From this information you can begin to define and confirm optimal epitopes for your antibody. This information is critical for antibody production and helpful when selecting commercial antibodies.
Know your antigen's biological characteristics
Understanding the biological relevance of your target, such as when, where, and which variant of your target is expressed, is valuable information when designing your experiment. This information can help identify potential non-specific interactions when validating your antibody.
For Example
A protein involved in the electron transport chain is likely to have a mitochondrial localization. Therefore, a nuclear staining pattern would be questionable. Databases (such as Uniprot, Genecards, AceView, Protein Atlas, or Expression Atlas) are particularly useful sites for gathering this type of information. See Additional Resources for a list of helpful resources.
Be cautious when evaluating resources based on mRNA expression, because protein and mRNA expression patterns do not always correlate.
Identify Cells and Tissues
A critical component of demonstrating antibody specificity Ability to recognize the target antigen. and selectivity Ability to preferably bind the target antigen in the presence of other antigens. is the correct use of controls. Determining which samples will be used for control materials is critical for the validation process.
Positive controls should confirm expression of your target antigen in the relevant cells and tissues.
Negative controls should consist of samples where the target is known to be absent, expressed at low levels, knocked down, or knocked out.
Positive and negative controls
Potential positive and negative controls can often be identified through online and literature searches. When selecting control samples, the expression of the target antigen should ideally be verified using more than one assay format, e.g., confirming expression by Western blot before use as an In-Cell Western Assay control.
It is best if one of these assays is a non-antibody method, such as genomic analysis. This increases confidence in the expression profile of the control samples, thereby enhancing the validation process. In addition to using tissues or cell lines that endogenously over- and under-express your target, both genetic and chemical approaches can be used to obtain valuable biological controls.
Genetic approaches for controls
Cultured cell lines are particularly beneficial when the target antigen expression can be altered by genetic modifications.
CRISPR/Cas9
This system offers an exacting method of gene editing. It can be used for complete genetic insertions and knockouts, as well as the insertion or correction of genetic mutations.
RNAi
Thoughtfully planned RNAi experiments, resulting in the absence or decrease of the specific target protein, are a useful tool for antibody validation. However, due to possible effects of off-target toxicity (10), care should be taken in how RNAi-based experiments are interpreted.
Chemical approaches for controls
In addition to genetic manipulation, chemical treatments with a well characterized substance (e.g., drug, growth hormone, or mineral) can be used to precisely alter protein expression in a controlled manner to obtain positive and negative controls.
For Example
Samples treated with and without a growth hormone, that stimulates the phosphorylation of a signaling protein, can be used as the positive and negative controls for an antibody targeting the phosphorylated antigen.
Positive controls with variable expression
Finally, selecting a few positive controls expressing differing amounts of the target antigen (e.g., high versus moderate or weak) can increase confidence in antibody specificity. These controls are helpful for selecting antibodies with the highest binding affinity Binding strength between the antibody’s binding site and its epitope. and highest specificity for the antigen.
Establish Experimental Prerequisites
Establish some preliminary assay parameters for the In‑Cell Western Assay prior to antibody selection to maximize the likelihood of antibody success.
The success of an antibody in one application does not predict its success in another application. Some antibodies will specifically recognize a fully denatured protein in a Western blot but will not recognize the same protein in its natural conformation in an immunoprecipitation experiment. Some of the assay parameters that may influence antibody performance and deserve consideration before antibody selection include:
Antibody/reagent compatibility
Live versus fixed cells
Fixation conditions
Permeabilization conditions
Blocking buffer constraints
Preferred normalization method
Determine Antibody Requirements
Examples of antibody requirements to consider include:
Polyclonal versus monoclonal versus recombinant antibodies
Direct versus indirect detection
Quantity of antibody for the project
Isotype
Host species
Polyclonal vs Monoclonal
Choosing between polyclonal and monoclonal antibodies can depend on antibody availability. If both options are available, you must decide if the technical advantages of a polyclonal antibody outweigh the disadvantages.
| Advantages | Disadvantages | |
| Monoclonal |
|
|
| Polyclonal |
|
|
Isotype
Selecting primary antibodies from different species or different isotypes is important when detecting multiple antigens in the same samples using multiple antibodies. These antibodies can subsequently be detected with individually labeled isotype-specific or species–specific secondary antibodies. Also, to avoid cross-reactivity of the secondary antibody, the species of the primary antibody should vary from that of your sample and blocking buffer.
Acquire Acceptable Antibodies
Once you have identified the fundamental requirements for the antibody, the next step is to source and compare available antibodies to find one that will meet the needs of your research. This section details the information that you should gather before purchasing antibodies to validate.
Identify Specific Antibodies
When reviewing antibodies, it is important to note the clone name or product number. This information is helpful when surveying scientific literature for successful antibody applications. Since there are often multiple suppliers of the same antibody, it can also prevent purchasing the same antibody from different suppliers. In addition to clone names and product codes, Research Resource Identifiers (RRID) are unique identifiers to label biological resources like antibodies. These identifiers can be obtained through the RRID Portal website (scicrunch.org) or by contacting the suppliers directly.
Review Literature
Antibody data sheets and scientific literature are typically the primary sources of information when researching antibodies. Peer-reviewed scientific literature is an excellent place to start for locating which antibodies have previously been used to study a target protein. Research articles can be found on PubMed, CiteAb, BenchSci, and commonly used search engines.
For some antigens, there is a clear choice of antibody based on supporting validation data. For other antibodies, there may be few antibody options and little supporting data available. Extensive validation may be required after purchase to ensure an antibody will work for you under your specific experimental conditions.
Locate Available Antibodies
An important consideration when prioritizing antibodies is whether or not they are easily available (not all are commercially available). Occasionally, research laboratories will choose not to share their reagents and supply of some polyclonal antibodies may be limited or no longer available.
In addition to searching individual company catalogs, there are several web-based search engines useful for identifying and comparing antibodies from commercial suppliers. These websites include Biocompare, The Antibody Resource, Antibodypedia, Linscott’s Directory, and the CPTAC Antibody Portal.
When planning a potentially lengthy research project, ensure you can acquire enough antibody to complete the project. This is particularly important with polyclonal antibodies. Keep in mind that researchers from other labs may have difficulty reproducing research conducted with polyclonal antibodies if antibodies from the same lot are no longer available. It may be worth validating the reagent and then purchasing multiple vials from the same lot to reduce difficulties with assay reproducibility.
Do not assume that antibody performance will be the same between lots. Each time you purchase antibodies from a new lot, validate and qualify antibodies from the new lot.
Obtain Antigen Information
The antigen used as an immunogen affects the number of possible antibody epitopes and impacts the antibody’s selectivity for the target protein. If a company does not readily supply this information, they may be willing to confirm if specific protein regions are contained within their immunogen without disclosing the actual immunogen.
If no information is available, assume the antibody may recognize any portion of the full-length protein.
Select Desired Host Species
Host species information is important for In-Cell Western Assay experiments that require multiple antibodies.
Antibodies raised in one species may recognize orthologous antigens in other species. Should you choose to use primary antibodies from the same species, you will need to find a way to avoid the problem of secondary antibodies binding to both primary antibodies.
Using primary antibodies raised in different species will avoid problems caused by secondary antibodies binding to both primary antibodies. It is also best to use affinity-purified secondary antibodies.
If you have questions about host species reactivity that is not listed on the antibody data sheet, contact the supplier.
Research Verified Applications
Antibody data sheets often include a list of validated techniques, suggested working dilutions, and additional technical information. Images from representative experiments are also often included to help you evaluate the data and draw your own conclusions. If an application is not listed, it may mean that the antibody has not been tested in that application. Some antibody suppliers will state when an antibody is not suited for a specific application, if evidence exists that shows the antibody does not work in that application.
For In-Cell Western Assays, antibodies validated for immunofluorescence, immunocytochemistry, and immunohistochemistry are sometimes a good place to start. It is the researcher’s responsibility to verify.
Compare Antibody Data to Existing Biological Information
As discussed previously, the biological information of the target is vital. Use this information to guide your selection and testing of an antibody. Compare the data found in the data sheet with known biological information (molecular weight, subcellular localization, tissue distribution, etc.).
For Example
If immunofluorescent staining revealed nuclear localization for a membrane receptor protein expected to be expressed at the plasma membrane, this would indicate that the antibody is non-specific.
If the data generated with one antibody differ from another antibody, further investigation is required to determine which antibody is selective for the target antigen.
Research the Supplier
When shopping for an antibody, there is less risk when you purchase from a supplier with a good reputation for customer satisfaction.
Notes About Custom Antibodies
Specific guidelines for producing and validating custom antibodies are beyond the scope of this guide, besides a brief mention. When producing custom antibodies, the type and quality of immunogen, host animal species, purification method, and pre-adsorption process should all be carefully considered.
Validate Antibody
Whether producing your own antibody or selecting from commercial offerings, it is important to validate antibody specificity and ensure that there is sufficient sensitivity across the entire dynamic range of samples to be tested in the In-Cell Western Assay.
Gather Antibody Validation Information
The antibody information in this section can likely be gleaned from published sources.
Reactivity to the immunogen
The following assays are often used to assess antibody reactivity against the immunogen:
Enzyme-linked immunosorbent assay (ELISA)
Flow cytometry
Western blotting
Antibody validation limited to an ELISA assay using the immunizing peptide alone is insufficient and does not guarantee reactivity with the endogenous protein.
Detection of a target antigen
Antibody reactivity with purified recombinant proteins or cells overexpressing the target protein are commonly used to demonstrate binding to the target antigen. Tagged expression constructs or lysates for most proteins are now commercially available to facilitate antibody validation. However, this still does not ensure antibody reactivity with the endogenous antigen.
Recognition of endogenous protein
Positive and negative controls are used to assess endogenous antibody binding. For thorough validation, multiple positive and negative controls should be used in the same experiment. Proper controls are essential for verification of endogenous protein detection and antibody specificity.
Antibody specificity and selectivity
Detection of the target antigen does not exclude the possibility that the antibody is cross-reactive with another protein(s). In addition to proteins sharing sequence similarity, antibodies can also recognize proteins with similar conformational epitopes, which cannot be predicted from amino acid sequence comparison. Antibody cross-reactivity with related proteins should be tested experimentally. A common approach is to screen transiently transfected cells engineered to express protein members of the same gene family.
Expression pattern
Experimentally confirm that the antibody detects the appropriate tissue and subcellular localization of the antigen.
Corroboration of multiple antibodies
Evaluating several antibodies in parallel and comparing their patterns of reactivity can significantly increase confidence in the antibodies. This is especially important when new or unexpected results are obtained.
Reagents
Some reagents work in a wide number of techniques whereas others are very limited in their application. Select an antibody that has been validated, when possible, in the same tissue type and species. If this information is absent, then additional experimental testing is advised.
Evidence for application suitability
An antibody might be validated in only a few techniques, but this does not suggest it is unsuitable for other applications. For example, an antibody recommended for IF may also work in the In-Cell Western Assay. Furthermore, many antibodies are validated for Western blotting, where a denatured antigen is usually recognized, and antibodies that selectively recognize the native conformation of the antigen may not work in this technique.
Validate Antibody Experimentally
For an In‑Cell Western Assay, antibody specificity should be evaluated by both Western blot and immunocytochemistry (ICC). Western blotting can yield useful information about antibody specificity, even though antigen presentation is different than in a cell-based assay. An antibody that generates any non-specific bands on a Western blot may indicate that the antibody is not suitable for In-Cell Western Assay experiments or that the blocking and antibody incubation conditions require further optimization.
Western blot analysis is a reliable way to determine if the desired cellular response has been triggered in your experiment. You may need to optimize cell treatment conditions or timing to capture the cellular response at its peak. For example, some phosphorylation events increase and decrease rapidly, so sampling at the wrong time may cause you to over or underestimate the response.
Determine necessary validation and optimization
If there is insufficient data to indicate that the antibody is suitably validated for your experimental context, then it is incumbent on the researcher to perform the additional required validation using appropriate controls.
Even if there is sufficient data to indicate that the antibody is suitably validated for your experimental context, then optimization is probably still required to achieve optimal performance of the antibody.
Western Blot Validation
Prepare your samples and perform the Western blot procedure
Follow LICORbio Protocols (licor.com/icw) for blocking, primary antibody incubation, secondary antibody incubation, wash steps, and image acquisition. Use LICORbio IRDye® Secondary Antibodies appropriate to the primary antibody you are evaluating.
Estimate total protein concentration, preferably by BCA, Bradford, or Lowry spectrophotometric assays, and load equal amounts of total protein onto the SDS-PAGE gel for each of the prepared lysate samples.
Whole cell lysates should be prepared from a representative cell line or cell type of interest. Recombinant proteins can also be used as a control.
Lysates should be prepared from cells treated under representative In-Cell Western Assay conditions.
Be sure to include a negative control lysate to detect any non-specific binding.
Include a high-quality protein molecular weight marker (licor.com/bio/reagents/protein-ladders) to verify target size. Follow standard protocols for gel electrophoresis and Western blot transfer.
Stain membrane with Revert™ 700 Total Protein Stain or Revert 520 Total Protein Stain (licor.com/revert) for total protein detection and normalization.
Verify Molecular Weight and Specificity
The position of the band(s) of interest on the Western blot should be close (e.g., ± 10% if using MW markers to size proteins) to the expected molecular weight of the target protein. Empiria Studio® Software (licor.com/empiria) provides analysis workflows to help with antibody validation.
Only one band should be visible in the lane of interest. If multiple bands are present – especially strong ones – the primary antibody is possibly cross-reactive and not suitable for use in the In-Cell Western Assay.
For activation-state-specific antibodies (e.g., phospho-specific antibodies), ensure that basal signal in the negative control lysate is not too high.
Comparing Antibody Selectivity and Specificity by Western Blot

Immunocytochemistry Validation
Antibody specificity and selectivity should also be evaluated in ICC to ensure optimal assay performance and interpretation. Signal can be assessed using multiwell plate assays on an Odyssey M, Odyssey DLx, or Odyssey CLx, or Odyssey F Imager, or by near-infrared fluorescence microscopy. General methods for specificity evaluation are described below. The primary antibody supplier may have additional suggestions or information about antibody specificity and optimization.
Primary antibody titration
Primary antibodies should be titrated to determine the concentration that provides optimal signal and lowest background. Careful titration may reduce background staining.
The multiwell format of the In‑Cell Western Assay makes it easy to quickly screen a range of antibody concentrations. Fluorescence microscopy should be used to confirm the protein localization at whichever primary antibody concentration is determined to be optimal.
Technical experimental conditions
If target signal is low, consider whether alternate experimental conditions (e.g., fixation, permeabilization, and blocking conditions) might improve performance.
Validation of primary antibodies in general
For validation of primary antibodies in general, create conditions under which the target signal increases or decreases (in each case, confirm the change in target signal by Western blotting or In‑Cell Western). Suggestions include:
Stain cells that express the target protein (endogenously and/or overexpressing), and cells that do not (endogenously or by knockout/knockdown). This is the most conclusive test. If this is not practical, look for cell lines that are high or low expressors of your target protein, or transfect the target into a cell line that does not endogenously express it.
Expose the cells to conditions, ligands, or inhibitors that affect target abundance.
Run antibody isotype controls (with Ig concentration and isotype that match your primary antibody) to assess non-specific binding.
Examine stained cells with a fluorescence microscope, to determine if target signal is localized to the proper cell compartment.
Validation of phospho-antibodies specifically
For validation of phospho-antibodies used to monitor cell signaling:
Treat fixed cells with phosphatase to reduce target phosphorylation. Signal should be reduced in treated cells.
This test cannot determine if the antibody cross-reacts with another phospho-epitope.
Treat cells with a target-specific ligand or inhibitor that modulates only the pathway of interest, to determine if signal responds accordingly. Confirm results by Western blotting.
Document and Report Antibody Details and Validation Procedures
Be sure to document your antibody details and validation procedure to ensure that all experimental details will be available for publication and to share with other researchers. Provide enough technical information to enable other researchers to acquire the same antibody and to get the same results from the same validation procedures and experiments. Empiria Studio® Software (licor.com/empiria) provides data fields in the antibody validation workflows that allow you to record this information. The Empiria Studio Antibody Validation Library provides a central location where you can find your antibody validation information. If allowed by your institution, share details about your antibody validation on online antibody databases (see Additional Resources).
Additional Resources
Single Antibody Titration Experiment
The following plate layout shows a template experiment that can be adapted for the specifics in your research to determine the best antibody dilution to use in your experiment. Plate layouts may also be created and printed from Empiria Studio Software.
The section Plate Loading Guides has loading guides designed to fit under a plate to help keep track of which samples should be loaded in which wells.

Plate Layout Legend
| Symbol | Definition |
| blnk | Blank wells contain only TBS or PBS buffer and are used to mitigate the potential impact of plate well edge effects. |
| bkg | Background wells that contain secondary antibody without primary antibody or normalization reagent. |
| + | Positive control wells contain primary and secondary antibody. Depending on your experiment, positive controls could be an over-expressed cell line, a treatment known to stimulate target expression, etc. |
| - | Negative control wells contain primary and secondary antibody. Depending on your experiment, negative controls could be a CRISPR knockdown, a treatment known to inhibit target expression, etc. |
Antibody Resources
European Monoclonal Antibodies Network
euromabnet.com"EuroMAbNet represents the first European network of laboratories linked to academic institutions, each having an internationally recognized reputation in the production and use of monoclonal antibodies."
CiteAb
citeab.comData provider for life sciences information.
The Antibody Registry
antibodyregistry.org"The Antibody Registry gives researchers a way to universally identify antibodies used in their research." You can find a list of LICORbio antibodies on The Antibody Registry by searching for "LI-COR".
The Biocompare Antibody Search Tool
biocompare.com/AntibodiesThe Biocompare Antibody Search Tool lets researchers search over 3 million antibodies from hundreds of antibody suppliers according to criteria such as target antigen, species reactivity, host, conjugate, isotype, and application.
Antibodypedia
antibodypedia.com"Antibodypedia scores antibodies to guide researchers to choose an appropriate antibody for a particular application."
RRID Portal
scicrunch.org/resourcesThe Research Resource Identifier (RRID) Portal provides shared identifiers for citing research resources in biomedical literature. You can add a resource and get a unique identifier if you notice that a resource you are using is missing.
Clustal Omega
clustal.org/omegaClustal Omega is a software tool that enables alignment of hundreds of thousands of sequences in a few hours.
Linscott’s Directory
linscottsdirectory.comSearch engine for finding suppliers for antibodies, ELISA assay kits, cytokines, enzymes, recombinant proteins, siRNAs, tissues, organs, antibody services, etc.
Expression Atlas
ebi.ac.uk/gxa/homeThe mission of the Expression Atlas is to provide the scientific community with freely available information on the abundance and localization of RNA and proteins for different species, biological conditions, tissues, cell types, developmental stages, disease state, and others.
AbMiner
discover.nci.nih.gov/abminer/home.do"AbMiner is a tool that allows users to search for appropriate, commercially available antibodies for research purposes, and to match each antibody to its respective genomic identifiers."
HUGO Gene Nomenclature Committee Search
This site enables you to search human gene nomenclature by symbol, keyword, and ID. The HGNC approves unique symbols and names for human loci.
National Center for Biotechnology Information
ncbi.nlm.nih.gov"The National Center for Biotechnology Information advances science and health by providing access to biomedical and genomic information."
UniProt
uniprot.org"The mission of UniProt is to provide the scientific community with a comprehensive, high-quality and freely accessible resource of protein sequence and functional information."
BenchSci
benchsci.comBenchSci provides reagent selection and experimental design aided by artificial intelligence.
GeneCards
genecards.org"GeneCards is a searchable, integrative database that provides comprehensive, user-friendly information on all annotated and predicted human genes."
Center for Strategic Scientific Initiatives Antibody Portal
proteomics.cancer.gov/antibody-portalThe CPTAC Antibody Portal serves as a National Cancer Institute (NCI) community resource that provides access to a large number of standardized renewable affinity reagents (to cancer-associated targets) and accompanying characterization data.
The Human Protein Atlas
proteinatlas.orgThe Human Protein Atlas is a program with the goal to map all the human proteins in cells, tissues, and organs using various omics approaches.
The Antibody Resource
antibodyresource.comSource for Antibody and ELISA products from major suppliers.
AceView
ncbi.nlm.nih.gov/IEB/Research/Acembly/"AceView provides a curated, comprehensive and non-redundant sequence representation of all public mRNA sequences..."
Antibody Selection References
10. Lin, A., Giuliano, C.J., Palladino, A., John, K.M., Abramowicz, C., Yuan, M.L., et al. (2019). Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Science Translational Medicine, 11(509). https://doi.org/10.1126/scitranslmed.aaw8412
11. Global Biological Standards Institute. Antibody Validation Standards, Policies, and Practices Workshop Report. 2016.
Fixation and Permeabilization
Fixation Chemically preserving biological specimens to limit degradation, prevent microbial contamination, and to adhere cell monolayers to a glass or plastic surface. and permeabilization Using solvents or detergents to create openings in the cell membrane so that antibodies and stains can gain access to intracellular targets. are tools that safeguard cell structure and allow entry of the antibody through the cellular and nuclear membranes. This section reviews common fixation and permeabilization methods, addresses experimental and technical considerations, and includes the two most common fixation/permeabilization protocols for In-Cell Western Assays.
Fixation Methods
Fixatives Chemicals that preserve biological specimens by limiting enzymatic degradation and preventing microbial contamination. preserve biological specimens by limiting enzymatic degradation, preventing microbial contamination, and fixing (cross-linking) cells to the plate. This is especially important for samples that have long lag times from collection to analysis. Stabilizing cellular architecture is another important benefit of fixation when using conditions too harsh for live cells or employing immunostaining techniques that have multiple solution changes.
There are a variety of strategies to fix cells, but the priority must be for preservation of antigenicity and epitope accessibility. The two groups of reagents most often used for fixation in cell biology, immunology, and medicine are aldehydes and organic solvents.
Recommendation: A 3.7% formaldehyde solution is the standard fixative when optimal fixation conditions are not known.
Aldehyde Fixatives
Aldehyde-based fixatives include formaldehyde, paraformaldehyde, and glutaraldehyde.
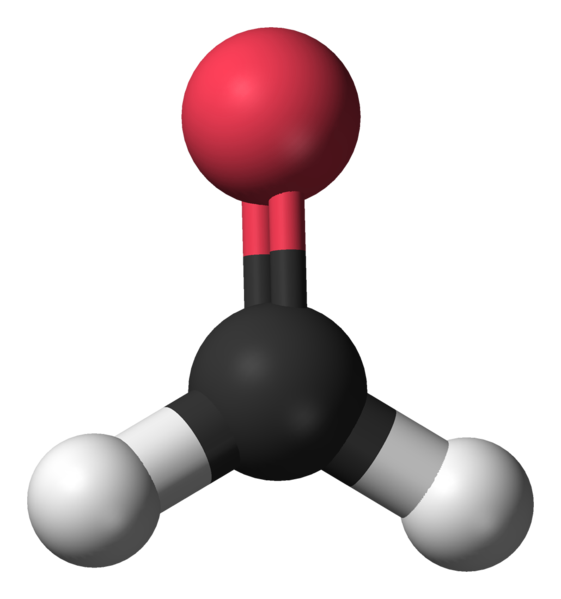
Formaldehyde and Paraformaldehyde
Formaldehyde (the saturated form in water at 37 - 40% is called formalin) is the most commonly used fixative that works for a wide range of cells and tissues, preserving the cellular structure without substantial cellular shrinkage (12 - 13). Formaldehyde is purchased as a saturated solution containing 10 - 15% methanol as a stabilizer to prevent repolymerization into paraformaldehyde or oxidation to formic acid. When diluted to an often-used concentration of 3.7 - 4%, the methanol concentration is 1 - 1.5%. This means that methanol is likely a factor in the permeabilization process and will be discussed further.
Paraformaldehyde is polymerized formaldehyde that is dissolved in heated (60 °C) water or PBS, and the pH is adjusted to 6.9. Paraformaldehyde does not contain added methanol. Formaldehyde and paraformaldehyde act by cross-linking functional groups of proteins, glycoproteins, nucleic acids, and polysaccharides.
{kind=link}
Glutaraldehyde
The bifunctional glutaraldehyde molecule has aldehyde groups at either end providing the advantage of greater cross-linking with the drawback of potential reduction in the immunogenicity of proteins for immunostaining (14).
{kind=link}
Combinations
Sometimes combinations of these reagents, such as formaldehyde and glutaraldehyde, are used to balance out the advantages and disadvantages of each one. Inclusion of glutaraldehyde, at a concentration between 0.05 – 0.1%, along with formaldehyde or paraformaldehyde may enhance the cross-linking action and thereby provide stronger cell fixation. The extent and nature of the crosslinking of any aldehyde can be controlled by the concentration, time, and temperature (see Choosing Fixation and Permeabilization Options).
Organic Solvents
Organic solvents adhere cells to surfaces by dehydration and precipitation of proteins. Methanol, acetone, and ethanol are commonly used and are very effective for nucleic acid preservation.
 |  |  |
{kind=link}
{kind=link}
{kind=link}
With the exception of methanol, other organic solvents can be significantly damaging to cell structure, cell organelles, microtubules, and nuclear content (15 - 16). Methanol can maintain protein cytoskeletal structure, although small molecules can be lost if not precipitated. This solvent often gives very good results for immunostaining with low background (17). Methanol fixation is frequently combined with an acetone rinse to improve permeabilization and overall results.
Evaluating Fixation
Optimizing fixation requires that you first decide if you will permeabilize the cells or not. The In-Cell Western Assay protocol calls for cell permeabilization. The On-Cell Western assay protocol is similar to the In-Cell Western Assay protocol, but cells are not permeabilized, enabling quantitative monitoring of cell surface protein expression.
Evaluating Fixation for In-Cell Western Assays
If you plan to permeabilize cells as you would in an In-Cell Western Assay, then try an aldehyde fixative followed with a permeabilization method or use an alcohol-based fixative at varying concentrations (Figure 116) on a single sample type to determine which method gives the best results.
Optimizing Fixation for On-Cell Western Assays
Because cells are not permeabilized in an On-Cell Western assay, you will need to choose paraformaldehyde or glutaraldehyde (Figure 116) for fixing cells. Other fixation methods will result in some degree of permeabilization. Evaluate various concentrations on a single sample type to determine if paraformaldehyde or glutaraldehyde is best for your experimental system.

Permeabilization
Fixation is commonly followed by a permeabilization step so that antibodies can gain access to the intracellular components. Permeabilization can be achieved with either detergents or solvents.
Detergents
Saponin is a relatively gentle detergent that removes cholesterol from the membrane, leaving holes to permit entry of antibodies (18).
Triton™ X-100 is a nonionic surfactant that is widely used, and its effects are non-specific and dependent on the amount and duration of exposure to the cells (19).
Recommendation: A 0.1% Triton X-100 solution is the standard permeabilization agent when optimal permeabilization conditions are not known.
Nonidet™ P-40 - Similar to Triton X-100, Nonidet P-40 is now available as IGEPAL CA-630. Nonidet P-40 is a non-specific, non-denaturing detergent that can have harsh effects on the nuclear and plasma membranes of cells if left for too long (18).
Tween® 20 - One of the gentlest nonionic detergents, Tween 20, can create pores without affecting the membrane integrity and is suitable for detection of cytoplasmic and nucleic acid targets in the cytoplasm (20).
Organic Solvents
Organic solvents can be used for permeabilization or both fixation and permeabilization combined. Two solvents frequently used together are methanol and acetone. Although they can be less effective and cause cell shrinkage, solvents do work well for nuclear targets.
Keep in mind that most 96- and 384-well plates are plastic and are not compatible with 100% acetone or chloroform fixatives. Only use glass bottom plates for those fixation solvents. For compatibility information, consult information provided by the plate manufacturer or ask the plate manufacturer for more information.
Optimize Permeabilization
Optimization of permeabilization requires that you decide on a single fixative protocol then vary the agents used to permeabilize the cell. The choice is dependent on the goal of your experiment. For example, cytoplasmic and nuclear targets will require gentler detergents (Tween 20, saponin) and careful timing. Small molecule targets may not be retained if methanol is used.
No single fixation/permeabilization method will perform best for every cell line or condition. Optimal conditions need to be empirically determined. Past experience with an antigen may be helpful, as are established conditions for this antigen for immunofluorescent microscopy or other cell-based assays. Primary antibody manufacturers will often provide fixation and permeabilization conditions for immunofluorescent staining of a particular antigen. Choosing Fixation and Permeabilization Options gives general concentration ranges, times, and temperatures to try for both fixative and permeabilization solutions. Optimization is the key to a quality, robust experiment.
Choosing Fixation and Permeabilization Options
| In-Cell Western | Pro | Con |
| Formaldehyde | Low level of cell shrinkage Good preservation of cell structure Wide range of cells and tissues | Always get a degree of permeabilization due to presence of methanol |
| Paraformaldehyde | Low level of cell shrinkage Good preservation of cell structure Wide range of cells and tissues | |
| Glutaraldehyde | Good for high MW DNA preservation | |
| Methanol | Preserves nucleic acid content High-level immunostaining with low background | |
| Acetone | Can only use 80% or lower on plastic plates | |
| Triton™ X-100 | Good at removing adducts Non-selective | |
| Tween® 20 | Gentle on cell lipids | |
| Saponin | Specifically interacts and removes cholesterol No significant membrane alteration | |
| NP-40 | Non-selective |
| Fixative | Permeabilization | Concentration Range |
Formaldehyde* | X | X | 2 - 3.7% in PBS |
Paraformaldehyde** | X | - | 4% in PBS |
| Glutaraldehyde | X | - | 2.5% in PBS |
| Methanol | X | X | 50 - 100% cold |
| Acetone | X | X | 70 - 100% cold |
| Triton™ X-100 | - | X | 0.1 - 0.2% |
| Tween® 20 | - | X | 0.1 - 0.5% |
| Saponin | - | X | 0.1 - 0.5% |
| NP-40 | - | X | 0.1 - 0.2% |
*37% (wt/vol) in H2O, contains 10 - 15% Methanol as stabilizer to prevent polymerization
** Paraformaldehyde is a methanol-free, stabilizer-free formaldehyde solution
On-Cell Westerns
If you are interested in cellular membrane targets rather than intracellular targets, then permeabilization is a step that you would either skip or shift to a later stage of the protocol. Your choice of washing buffer would not include detergents but would include buffers as close to physiological pH as possible, such as PBS or cell culture media.
If you intend to normalize cell number using a cell impermeant stain, such as CellTag™ 700 Stain, then prior to the addition of the secondary antibody step, you would need to add a permeabilization step to allow entry of CellTag 700 Stain. The stain would be mixed with the secondary antibody and added at the same time. The secondary antibody will only recognize the primary antibody that has already bound to the cell membrane proteins. Below is an example of a generic On-Cell Western workflow.
Fix cells with paraformaldehyde or glutaraldehyde.
Wash with PBS or cell culture media.
Block cells.
Add primary antibody.
Wash with Triton™ X-100 / PBS.
Add secondary antibody and cell normalization stain.
Wash with Triton X-100 / PBS.
Image
Recommended Fixation and Permeabilization Methods
Method 1: Formaldehyde/Triton™ X-100
The following conditions are a good place to begin optimization if you have little or no experience with your system. In this method, formaldehyde is used as the cross-linking reagent, and Triton X-100 is used for permeabilization.
Just prior to use, prepare Fixation Solution:
Prepare 20 mL per plate.
Mix 2 mL 37% Formaldehyde and 18 mL 1X PBS (final formaldehyde concentration = 3.7%)
Formaldehyde is hazardous and should be handled and discarded according to the manufacturer’s MSDS.
Carefully remove growth medium from the microwell plate by manual aspiration.
Immediately fix cells. Using a multichannel pipette, carefully add 150 µL of Fixation Solution down the sides of each well to avoid detaching cells.
Cover the plate and allow incubation on the bench top for 20 minutes at ambient temperature without agitation.
During the fixing step, prepare Permeabilization Solution by mixing 200µL of 10% Triton X-100 and 19.8 mL of 1X PBS (final Triton X-100 concentration = 0.1%).
Remove Fixation Solution from the microwell plate by manual aspiration to an appropriate hazardous waste container (refer to manufacturer’s SDS).
Using a multichannel pipette, carefully add 150 µL of Permeabilization Solution down the sides of each well to avoid detaching cells.
Gently agitate plate for 20 minutes at room temperature.
Remove Permeabilization Solution carefully from the wells using the multichannel pipette without removing any cells.
After removing the permeabilization wash by manual aspiration, proceed immediately to blocking and the remaining In-Cell Western incubation steps.
Method 2: Methanol
The second method uses methanol for both fixation and permeabilization of cells. Methanol fixation/permeabilization may be a better option than formaldehyde in some cases (for example, when using some phospho-Stat3 antibodies or antibodies against some major structural proteins or nuclear matrix proteins).
Use 100% methanol that has already been cooled to -20 ˚C.
Methanol is flammable and should only be placed in a freezer that is appropriately rated for flammable liquids.
Carefully remove growth medium from the microplate by manual aspiration.
Immediately fix and permeabilize cells by adding 150 µL cold methanol to each well.
Be sure to carefully add the methanol down the sides of the wells to avoid detaching the cells from the well bottom.
Allow the plate to incubate at -20 ˚C for 10 minutes without shaking.
Carefully remove the methanol by manual aspiration into an appropriate waste container.
Gently rinse wells three times with 100 µL of 1X PBS. Be sure to add and remove the PBS carefully by manual aspiration to avoid detaching the cells.
After removing the last 1X PBS rinse, proceed immediately to blocking and remaining In-Cell Western incubation steps.
Method Comparison Example
The graph in Figure 117 shows the Z’-factor results when comparing permeabilization Method 1 and Method 2 for detection of Stat3 phosphorylation in EGF-treated and untreated A431 cells. In this case, methanol produced consistently more desirable results than formaldehyde and Triton™ X-100 (Z’-factor values > 0.5 are desirable).
Z’-Factor Analysis of Two Independent Fixation and Permeabilization Techniques

Alternative Methods
The following reagents/methods are alternatives to the aforementioned fixation/permeabilization methods. Because these methods have not been specifically tested for the In-Cell Western Assay, they are not supported by LI-COR. However, they are relatively common to other immunocytochemical assays, and may be viable options when optimizing your assay.
The steps in these methods are not rigid, and optimization of the various methods is recommended. As a general rule, aldehydes must be followed by a permeabilization step, but alcohols and organic solvents do not require a separate permeabilization step; however, this is not always true. In each case, proceed with In-Cell Western blocking and incubation steps after the fixation/permeabilization procedure.
80% Acetone in Water (100% acetone not compatible with plastic plates)
Consult your manufacturer’s documentation for specifics.
Fix cells with ice-cold (-20 ˚C) 80% acetone for 10 minutes.
Rinse with 1X PBS.
Formaldehyde-Methanol
Fix cells with room temperature 3.7% formaldehyde for 20 minutes.
Incubate cells with cold 100% methanol for 10 min.
Rinse with 1X PBS.
Ethanol-Acetic Acid
Fix cells with refrigerated (4 ˚C) 95% ethanol + 5% glacial acetic acid for 10 minutes.
Rinse with 1X PBS.
Methanol-Acetone (100% acetone not compatible with plastic plates)
Fix cells with methanol for 10 minutes at -20 ˚C.
Remove methanol and add acetone (-20 ˚C) for 1 minute.
Rinse with 1X PBS.
Paraformaldehyde
Fix cells in 4% paraformaldehyde for 20 minutes at room temperature.
Rinse with 1X PBS.
Wash 1 time for 20 minutes with 0.1% Triton™ X-100 (diluted in 1X PBS).
Paraformaldehyde-Methanol
Fix cells in 4% paraformaldehyde for 20 minutes at room temperature.
Rinse with 1X PBS.
Add methanol (-20 ˚C) for 5 minutes.
Rinse with 1X PBS.
Fixation and Permeabilization References
12. Thavarajah, R., Mudimbaimannar, V.K., Elizabeth, J., Rao, U.K., and Rangathan, K. (2012). Chemical and physical basics of routine formaldehyde fixation. J Oral Maxillofac Pathol, 16(2), 400-405. https://doi.org/10.4103/0973-029x.102496
13. Srinivasan, M., Sedmak, D., and Jewell, S. (2002). Effect of fixatives and tissue processing on the content and integrity of nucleic acids. Am J Pathol, 161(6), 1961-1971. https://doi.org/10.1016/s0002-9440(10)64472-0
14. Suthipintawong, C., Leong, A.S., and Vinyuvat, S. (1996). Immunostaining of cell preparations: a comparative evaluation of common fixatives and protocols. Diagn Cytopathol, 15(2), 167-174. https://doi.org/10.1002/(sici)1097-0339(199608)15:2%3C167::aid-dc17%3E3.0.co;2-f
15. Hoetelmans, R.W., Prins, F.A., Cornelese-ten Velde, I., van der Meer, J., Van de Velde, C.J., and van Dierendonck, J.H. (2001). Effects of acetone, methanol, or paraformaldehyde on cellular structure, visualized by reflection contrast microscopy and transmission and scanning electron microscopy. Appl Immunohistochem Mol Morphol, 9(4), 346-351. https://doi.org/10.1097/00129039-200112000-00010
16. Vielkind, U. and Swierenga, S.H. (1989). A simple fixation procedure for immunofluorescent detection of different cytoskeletal components within the same cell. Histochemistry, 91(1), 81-88. https://doi.org/10.1007/bf00501916
17. Levitt, D. and King, M. (1987). Methanol fixation permits flow cytometric analysis of immunofluorescent stained intracellular antigens. J Immunol Methods, 96(2), 233-237. https://doi.org/10.1016/0022-1759(87)90319-x
18. Amidzadeh, Z., Behbahani, A.B., Erfani, N., Sharifzadeh, S., Ranjbaran, R., Moezi, L., et al. (2014). Assessment of different permeabilization methods of minimizing damage to the adherent cells for detection of intracellular RNA by flow cytometry. Avicenna J Med Biotechnol, 6(1), 38-46. http://www.ncbi.nlm.nih.gov/pmc/articles/pmc3895578/
19. Oliver, C. and Jamur, M.C. (2010). Immunocytochemical Methods and Protocols. Methods Mol Biol, 588, 63-66. https://doi.org/10.1007/978-1-59745-324-0
20. le Maire, M., Champeil, P., and Moller, J.V. (2000). Interaction of membrane proteins and lipids with solubilizing detergents. Biochim Biophys Acta, 1508(1-2), 86-111. https://doi.org/10.1016/s0304-4157(00)00010-1
Example Fixation and Permeabilization Optimization Plate Layout

Blocking Buffer Evaluation
Blocking is an important step in cell-based assay protocols. Once cells in an In-Cell Western Assay have been fixed and permeabilized, blocking buffer A solution (often consisting of proteins) that reduces nonspecific antibody binding by binding to areas in a well that are not occupied by cells and by binding to nonspecific interaction sites in/on cells. is added to bind spaces in the well not occupied by cells and to bind potential nonspecific binding sites in the cell. Blocking buffers minimize non-specific binding of primary and secondary antibodies to wells and to non-target sites in the cell.
Non-specific antibody binding can mask signal from the target protein, reducing specificity and sensitivity. Blocking allows detection of specific antibody binding to the target. Ultimately, blocking improves the signal-to-noise ratio by reducing background. High background can be a problem with immunoassays, so blocking is an especially important step that should always be performed.
No single blocking buffer is ideal for every target of interest. A blocking buffer used for one application may not necessarily be optimal for other applications. This section provides essential knowledge for choosing and optimizing a blocking buffer system for immunoassays and shows an experimental design that can be used to help identify the optimal buffer for an immunoassay.
Buffer System
Immunoassay protocols require solutions for blocking, antibody dilution, and washing.
PBS (Phosphate-buffered saline) and TBS (Tris-buffered saline) are commonly used buffers during the various stages in immunoassays, and it is useful to know when to use each of them. The buffer you choose will depend on your proteins of interest and the antibodies used. It is recommended that you compare PBS and TBS buffers to see which gives you the highest signal and lowest background for your specific targets.
PBS and TBS are often interchangeable as dilution buffers for blocking. However, not all blocking buffer formulations are suitable for every situation or antibody, and there are instances where PBS cannot be used.
TBS blocking buffers are typically preferred when detecting post-translational modifications such as phosphorylation. In some cases, the primary antibody may not only bind to the phosphate on the protein of interest, but also to phosphate in a PBS-based buffer, significantly reducing your target signal. However, this is not the case for all phospho-specific antibodies. For example, some PBS-based buffers can reduce the weaker non-specific interactions of a phospho-specific antibody while leaving the stronger antibody-antigen interactions intact. Therefore, both buffers need to be tested.
Blocking Agents
The most common blocking agents for cell-based assays include concentrated protein solutions and pre-formulated commercial solutions.
No single blocking buffer is ideal for every target of interest. It is important to optimize the blocking conditions for your cell line and protein target and determine which are most appropriate for the context and goals of your experiment. A good place to start is to look at published IF/ICC experiments where the same cell type and/or protein of interest has been examined and see what blocking conditions were used. This can give you some confidence as to which condition may work for your experiments.
Bovine Serum Albumin
Bovine serum albumin (BSA), derived from cow serum, can be used when detecting post-translational modifications. Because it is made of a single purified protein, BSA is less likely to interfere with antibody binding. Gelatin is also a purified protein, hydrolyzed collagen, that can come from different sources. Fish gelatin does not react with mammalian proteins and can decrease background in immunoassays, but may mask some antigens and should be carefully tested prior to use in immunoassays.
Synthetic Blocking Buffers
Synthetic and/or non-animal blocking agents were formulated to prevent any cross reactivity with the primary and secondary antibodies produced in animals. Pre-formulated commercial solutions include those produced by LI-COR. LI-COR offers several blocking buffers for use when performing an immunoassay.
Intercept® Blocking Buffer products are optimized for use with IRDye® products and other near-infrared fluorophores. They provide excellent performance with low variability and background for immunoassays. Intercept Blocking Buffer products are non-mammalian and provide a high signal-to-noise ratio in a 1X ready-to-use formula. Intercept Protein-Free Blocking Buffers, available in both PBS and TBS formulations, are essential for applications where animal-based products are prohibited, or protein-based blocking buffers are contributing to cross-reactivity.
Normal Serum
Normal serum is a common blocking agent that helps reduce the nonspecific binding of the primary and secondary antibodies to the cellular proteins by binding to cross-reactive sites. Serum is rich in antibodies, albumin, and other proteins that readily bind to nonspecific protein-binding sites which can improve the signal-to-noise of the assay. The most important factor to remember is the serum should be from the same host species as the secondary antibody. For example, if the primary antibody is a rabbit anti-target and the secondary is a labeled goat anti-rabbit, then the blocking serum would be normal goat serum (NGS).
Blocking Buffer Optimization

Z’-Factor Determination
During In‑Cell Western Assay development, it is important to assess the overall quality and reliability of the assay. The Z’-factor statistic provides a way to evaluate whether or not assay conditions (reagents, protocols, instrumentation, kinetics, and other conditions not directly related to the test compounds) are optimized. Z’-factor, introduced by Zhang et al., is a dimensionless value that represents both the variability and the dynamic range between two sets of sample control data.
Z’-factor experiments are performed on one or more In‑Cell Western Assay plates containing replicate wells designated for background subtraction, negative control samples, and positive control samples. A Z’-factor assay can be performed at any point during the assay development process.
Experimental Overview
As with any statistical parameter, the accuracy of the Z’-factor value improves with larger data sets used for the calculation (see Replicates). For a quality assessment, 3 - 4 plates should be run on different days. Wells can be arranged as desired, but a typical arrangement may look like the one shown in Figure 120.

Guidelines for the Z'-Factor Experiment
All reagents (apart from test compounds and reagents), antibodies, materials, and assay conditions should be the same as those that you intend to use for the final In‑Cell Western Assay.
Background wells must be included on the assay plate to account for nonspecific secondary antibody binding (see the Experimental Controls).
Be sure to correct for well-to-well variation by normalizing (see the Normalization).
When choosing treatment compounds to be used for Z’-factor analysis (i.e., stimulants, inhibitors, drugs, etc.), select well-characterized agents which give the best known response in an appropriate cell line.
Z'-Factor Calculation Overview
The Z'-factor value can be calculated manually from normalized signal intensity values, or you can use the Z'-factor calculation in Empiria Studio Software or Image Studio™ Software.
Z'-Factor Equation
This is the equation for calculating the Z'-factor and a list of the terms in the equation.

Where:
 = Standard deviation of positive controls
= Standard deviation of positive controls = Standard deviation of negative controls
= Standard deviation of negative controls = Mean of positive controls
= Mean of positive controls = Mean of negative controls
= Mean of negative controls
Z'-Factor Interpretation
The Z'-factor equation can also be written as follows.

Where:
 represents the assay dynamic range
represents the assay dynamic range is the separation band between positive and negative control signals.
is the separation band between positive and negative control signals.
Z'-factor values are always 1 or less as described.

Z’ = 1 Indicates an ideal assay. As standard deviations become very small or the difference between signals for positive and negative controls approaches infinity, Z’-factor approaches 1.
1 > Z’ ≥ 0.5 Indicates a high-quality assay exhibiting a wide separation between signal and background, and low data variability.
0.5 > Z’ > 0 Indicates a poor-quality assay with marginal distinction between signal and background, and higher data variability.
Z’ ≤ 0 Indicates unreliable data. Assay conditions are not optimized or the assay is not capable of generating meaningful data.
Z’ = -1 There is no distinguishable difference between background signal and sample signal. If the lower limit of detection (LLD) is expressed as the mean of the background signal plus three SD of the background signal, then the measured signal is equal to the LLD of the instrument.
Z'-Factor Summary
The Z’-factor experimental guidelines described above can be used to evaluate In-Cell Western Assay quality at any time throughout assay development, or near completion of assay optimization. Z’-factor measurement is also useful for monitoring assay quality over time, from user to user, or when conditions or reagents within the assay must be changed.